OpenFold: Retraining AlphaFold2 yields new insights into its learning mechanisms and capacity for generalization
Abstract
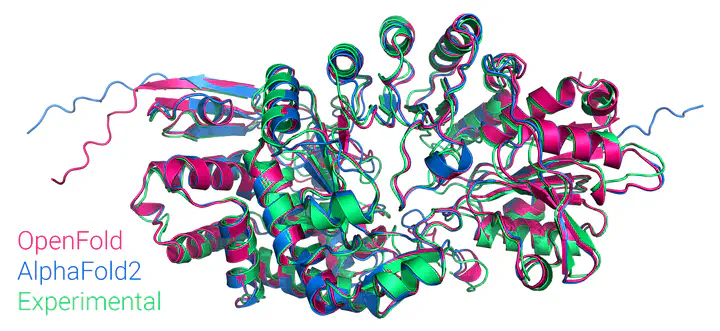
AlphaFold2 revolutionized structural biology with the ability to predict protein structures with exceptionally high accuracy. Its implementation, however, lacks the code and data required to train new models. These are necessary to (1) tackle new tasks, like protein–ligand complex structure prediction, (2) investigate the process by which the model learns and (3) assess the model’s capacity to generalize to unseen regions of fold space. Here we report OpenFold, a fast, memory efficient and trainable implementation of AlphaFold2. We train OpenFold from scratch, matching the accuracy of AlphaFold2. Having established parity, we find that OpenFold is remarkably robust at generalizing even when the size and diversity of its training set is deliberately limited, including near-complete elisions of classes of secondary structure elements. By analyzing intermediate structures produced during training, we also gain insights into the hierarchical manner in which OpenFold learns to fold. In sum, our studies demonstrate the power and utility of OpenFold, which we believe will prove to be a crucial resource for the protein modeling community.